
Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.
Smegenų lizencefalija
Medicinos ekspertas

Paskutinį kartą peržiūrėta: 12.07.2025
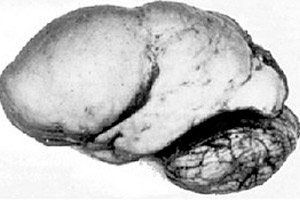
Tarp organinių smegenų patologijų išsiskiria tokia įgimta smegenų vystymosi anomalija kaip lisencefalija, kurios esmė slypi beveik lygaus pusrutulių žievės paviršiaus – su nepakankamu susisukimų ir vagų skaičiumi. [ 1 ]
Jei visiško susisukimų nėra, apibrėžiama agyrija, o kelių plačių plokščių susisukimų buvimas vadinamas pachigirija. Šie defektai, kaip ir kai kurios kitos smegenų redukcinės deformacijos, TLK-10 klasifikacijoje turi kodą Q04.3.
Epidemiologija
Remiantis retų ligų statistika, 100 tūkstančių naujagimių tenka 1–1,2 lisencefalijos atvejo. [ 2 ], [ 3 ]
Remiantis kai kuriais duomenimis, iki 25–30 % klasikinės lisencefalijos atvejų stebima vaikams, sergantiems Millerio-Dikerio sindromu; beveik 85 % pacientų aptinkamos LIS1 ir DCX genų taškinės mutacijos ir delecijos [ 4 ].
Genetiniai 17 su lisencefalija susijusių genų tyrimai parodė, kad LIS1 mutacija arba delecija lemia 40 % pacientų, o 23 % – su DCX mutacija, po to seka TUBA1A (5 %) ir DYNC1H1 (3 %).[ 5 ]
Priežastys lissencephaly
Visos žinomos smegenų žievės (cortex cerebri) susidarymo beveik arba visiškai be susisukimų ir vagų, kurios padidina žmogaus smegenų „darbo plotą“ ir užtikrina centrinės nervų sistemos „produktyvumą“, priežastys yra susijusios su jos perinatalinio vystymosi sutrikimais. Tai yra, vaisiui išsivysto lisencefalija. [ 6 ]
Vaisiaus smegenų žievės sluoksnių nesuformavimas sergant lissencephalija yra nenormalios ją formuojančių neuronų migracijos arba priešlaikinio šio proceso nutraukimo rezultatas.
Šis procesas, kuris yra būtinas smegenų žievės histogenezei, vyksta keliais etapais nuo 7 iki 18 nėštumo savaitės. Atsižvelgiant į padidėjusį jautrumą genetinėms mutacijoms, taip pat į įvairius neigiamus fizinius, cheminius ir biologinius poveikius, bet koks nukrypimas nuo normos gali lemti neteisingą neuronų lokalizaciją ir galimą sustorėjusio žievės pilkosios medžiagos sluoksnio, neturinčio būdingos struktūros, susidarymą. [ 7 ]
Kai kuriais atvejais vaikų lissencephalija yra susijusi su Miller-Diekerio, Walker-Warburgo arba Norman-Robertso sindromais.
Taip pat skaitykite – Smegenų vystymosi defektai
Rizikos veiksniai
Be kai kurių genų mutacijų, rizikos veiksniai, lemiantys vaiko, turinčio tokį sunkų defektą, gimimą, yra vaisiaus deguonies badas (hipoksija); nepakankamas kraujo tiekimas į smegenis (hipoperfuzija); ūminis smegenų kraujotakos sutrikimas perinatalinio insulto forma; placentos patologijos; nėščiosios virusinės infekcijos (įskaitant TORCH); [ 8 ] bendros medžiagų apykaitos ir skydliaukės funkcijos sutrikimai; rūkymas, alkoholis, psichotropinės ir narkotinės medžiagos; daugelio vaistų vartojimas; padidėjęs radiacijos lygis. [ 9 ]
Pathogenesis
Ne visais lissencephalijos atvejais patogenezę sukelia chromosomų anomalijos ir genų mutacijos. Tačiau yra žinomi kai kurie genai, koduojantys baltymus, kurie atlieka svarbų vaidmenį teisingame neuroblastų ir neuronų judėjime išilgai radialinių glijos ląstelių – formuojant smegenų žievę. O šių genų mutacijos sukelia šią patologiją. [ 10 ]
Visų pirma, tai yra sporadinės LIS1 geno mutacijos (be paveldimumo) 17 chromosomoje, kuri reguliuoja mikrovamzdelių citoplazminį motorinį baltymą dyneiną, taip pat DCX geno mutacijos X chromosomoje, kuri koduoja baltymą doublekortiną (lissencephaliną-X). [ 11 ]. Pirmuoju atveju specialistai apibrėžia klasikinę lissencephaliją (I tipo), antruoju – su X chromosoma susijusią. [ 12 ]
Kai FLN1 genas, koduojantis fosfoproteiną filaminą 1, yra pašalinamas, nukreiptos neuronų migracijos procesas gali iš viso neprasidėti, todėl visiškai išnyksta susisukimai (agirija). [ 13 ]
CDK5 gene, kuris koduoja kinazės fermentą – ląstelės viduje vykstančios medžiagų apykaitos katalizatorių, reguliuojantį CNS neuronų ląstelių ciklą ir užtikrinantį normalią jų migraciją prenatalinio smegenų struktūrų formavimosi metu, nustatytos mutacijos.
Nenormalūs RELN geno pokyčiai 7 chromosomoje, sukeliantys žievės girinius defektus Normano-Robertso sindrome, lemia ekstraląstelinio glikoproteino reelino, kuris yra būtinas nervinių kamieninių ląstelių migracijai ir padėčiai reguliuoti smegenų žievės vystymosi metu, trūkumą.[ 14 ], [ 15 ], [ 16 ]
ARX genas koduoja nearistaleninį homeobox baltymą – transkripcijos faktorių, kuris atlieka svarbų vaidmenį kaktinėse smegenyse ir kituose audiniuose.[ 17 ] Vaikams, turintiems ARX mutaciją, pasireiškia ir kiti simptomai, pavyzdžiui, trūkstamos smegenų dalys (jungtinio kūno agenezė), nenormalūs lytiniai organai ir sunki epilepsija.[ 18 ],[ 19 ]
Su lisencefalija susieti keli genai. Šie genai yra VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A ir CDK5.[ 20 ]
Citomegalovirusas (CMV) buvo susijęs su lisencefalijos išsivystymu dėl sumažėjusio kraujo tiekimo į vaisiaus smegenis. CMV infekcijos sunkumas priklauso nuo nėštumo amžiaus. Ankstyva infekcija labiau linkusi sukelti lisencefaliją, nes neuronų migracija vyksta ankstyvuoju nėštumo laikotarpiu.[ 21 ]
Be to, šios anomalijos atsiradimo mechanizmas apima nepilną arba vėlyvą neuronų migracijos iš periventrikulinės generatyvinės zonos į smegenų žievę nutraukimą. Tokiais atvejais išsivysto arba nepilna lissencephalija, arba pachigyrija, kurioje susidaro keli platūs grioveliai ir vingiuotės (tačiau dauguma jų nėra).
Simptomai lissencephaly
Pirmieji šios patologijos požymiai (nesant anksčiau minėtų sindromų) gali pasireikšti ne iš karto po gimimo, o po pusantro ar dviejų mėnesių. Dažniausiai pastebimi šie klinikiniai lissencepalijos simptomai:
- raumenų hipotonija, dažnai derinama su spazminiu paralyžiumi;
- traukuliai ir generalizuoti toniniai-kloniniai traukuliai (opistotonuso pavidalu);
- sunkus protinis atsilikimas ir augimo sulėtėjimas;
- Neurologinių ir motorinių funkcijų sutrikimas.
Rijimo problemos apsunkina kūdikio maitinimą. [ 22 ]
Didelis neuromotorikos sutrikimas dažnai pasireiškia tetraplegija – visų galūnių paralyžiumi. Galima rankų, pirštų ar kojų pirštų deformacija.
Normano-Roberto sindromo su I tipo lissencephalija metu stebimos kaukolės ir veido anomalijos: sunki mikrocefalija, žemas kaktos nuolydis ir išsikišęs platus nosies tiltelis, plačiai išdėstytos akys (hiperterlorizmas), žandikaulių neišsivystymas (mikrognatija). [ 23 ]
Millerio-Dikerio sindromui taip pat gali būti būdingas neįprastai mažas galvos dydis su plačia, aukšta kakta ir trumpa nosimi, įdubimai smilkiniuose (bitemporinės įdubos) ir žemai išsidėsčiusios, deformuotos ausys.
Sunkus lissencephalijos sindromas pasižymi mikrocefalija, akių obuolių dydžio sumažėjimu (mikroftalmija), kartu su tinklainės displazija, obstrukcine hidrocefalija ir smegenų jungties nebuvimu arba hipoplastiniu korpusu.
Komplikacijos ir pasekmės
Tarp šios anomalijos komplikacijų specialistai įvardija rijimo funkcijos (disfagijos) ir gastroezofaginio refliukso pažeidimą; refrakterinę (nekontroliuojamą) epilepsiją; dažnas viršutinių kvėpavimo takų infekcijas; pneumoniją (įskaitant lėtinę aspiraciją).
Kūdikiai, sergantys lisencefalija, gali turėti įgimtų organinių širdies problemų, pasireiškiančių prieširdžių pertvaros defektu arba kompleksiniu širdies defektu su cianoze (Fallot tetralogija). [ 24 ]
Pogimdyminio augimo sulėtėjimo pasekmės daugeliu atvejų baigiasi mirtimi per 24 mėnesius po gimimo.
Diagnostika lissencephaly
Diagnozė prasideda nuo vaiko fizinės apžiūros, tėvų ligos istorijos tyrimo ir nėštumo bei gimdymo istorijos.
Nėštumo metu gali prireikti atlikti beląstelinį vaisiaus DNR tyrimą, amniocentezę arba chorioninio gaurelio mėginių ėmimą. [ 25 ] Daugiau informacijos žr. – Įgimtų ligų prenatalinė diagnostika.
Instrumentinė diagnostika naudojama smegenų struktūroms vizualizuoti ir jų funkcijoms įvertinti:
- smegenų kompiuterinė tomografija;
- smegenų magnetinio rezonanso tomografija (MRT);
- elektroencefalograma (EEG). [ 26 ]
Nėštumo metu, po 20–21 savaitės, atliekant vaisiaus ultragarsinį tyrimą, galima įtarti lissencephaliją, nes nėra parieto-pakaušio ir kalcininių vagų bei yra smegenų Silvijaus plyšio anomalija.
Diferencialinė diagnostika
Atliekama diferencinė diagnostika su kitais įgimtų smegenų defektų sindromais.
Yra daugiau nei 20 lissencephalijos tipų, kurių dauguma skirstomi į 2 pagrindines kategorijas: klasikinę lissencephaliją (1 tipas) ir akmeninę lissencephaliją (2 tipas). Kiekviena kategorija turi panašius klinikinius požymius, bet skirtingas genetines mutacijas.[ 27 ]
Smegenų tyrimas sergant I tipo lissencephalija rodo keturių, o ne šešių sluoksnių smegenų žievę, kaip normaliems pacientams, tuo tarpu sergant II tipo lissencephalija smegenų žievė yra dezorganizuota ir atrodo gumbuota arba mazguota dėl visiško smegenų žievės išstūmimo žievės neuronų sankaupomis, atskirtomis gliomezenchiminiu audiniu. Pacientams taip pat buvo nustatyti raumenų ir akių sutrikimai.
- Klasikinė lissencephalija (1 tipas):
- LIS1: Izoliuota lissencephalija ir Millerio-Dikerio sindromas (lissencephalija, susijusi su veido dismorfizmu). [ 28 ]
- LISX1: DCX geno mutacija. Palyginti su LIS1 mutacijų sukelta lisencefalija, DCX žievė turi šešių, o ne keturių sluoksnių.
- Izoliuota lissencephalija be kitų žinomų genetinių defektų
- Akmenimis grįsta lissencephalija (2 tipas):
- Walkerio-Varburgo sindromas
- Fukujamos sindromas
- Raumenų, akių ir smegenų ligos
- Kitų tipų negalima priskirti nė vienai iš dviejų aukščiau išvardytų grupių:
- LIS2: Normano-Roberto sindromas, panašus į I tipo lissencephaliją arba Millerio-Dikerio sindromą, bet be 17 chromosomos ištrynimo.
- LIS3
- LISX2
Mikrolisencefalija: tai normalios žievės sulankstymo nebuvimo ir neįprastai mažos galvos derinys. Vaikai, kuriems pasireiškia įprasta lisencefalija, gimsta su normalaus dydžio galvomis. Vaikams, kurių galvos dydis gimsta su maža, paprastai diagnozuojama mikrolisencefalija.
Taip pat svarbu atskirti lissencephaliją ir polimikrogriją – skirtingus smegenų vystymosi defektus.
Su kuo susisiekti?
Gydymas lissencephaly
Lisencefalija yra neišgydomas organinis defektas, todėl galimas tik palaikomasis ir simptominis gydymas. [ 29 ]
Visų pirma, tai yra prieštraukulinių ir priešepilepsinių vaistų vartojimas, taip pat gastrostominio vamzdelio įvedimas į skrandį (jei vaikas negali savarankiškai nuryti). Masažas yra naudingas.
Esant sunkiam hidrocefalijos etapui, pašalinamas smegenų skystis.
Prevencija
Ekspertai rekomenduoja būsimiems tėvams kreiptis į genetines konsultacijas, o nėščiosioms laiku užsiregistruoti pas akušerius-ginekologus ir atlikti visus planinius tyrimus.
Prognozė
Vaikams, sergantiems lisencefalija, prognozė priklauso nuo jos laipsnio, tačiau dažniausiai vaiko protinis vystymasis neviršija keturių–penkių mėnesių lygio. Ir visi vaikai, kuriems diagnozuota ši diagnozė, kenčia nuo sunkių psichomotorinių sutrikimų ir sunkiai gydomos epilepsijos. [ 30 ]
Pasak NINDS (Amerikos nacionalinio neurologinių sutrikimų ir insulto instituto), maksimali gyvenimo trukmė žmonėms, sergantiems lissencephalija, yra apie 10 metų.